

2018年第七届中国罕见病高峰论坛在上海举行
“创新为源、患者至上”,夏尔携手多方共促中国罕见病事业发展
9月14-16日第七届中国罕见病高峰论坛于在上海成功举办。 本届论坛由上海四叶草罕见病家庭关爱中心与复旦大学附属儿科医院(国家儿童医学中心)联合主办,是迄今国内罕见病领域规模最大的综合性论坛,夏尔作为重要合作伙伴支持并参与其中。
为应对中国罕见病领域面临的诸多挑战, 夏尔在今年的高峰论坛上举办一场卫星会议和四场分组会议,并邀请多位专家发表精彩演讲。包括国内外罕见病领域的专家学者、临床医生、罕见病患者组织、患者家庭等700余名行业相关人士出席本次会议,围绕“创新与合作——让爱不罕见”的大会主题,共同探讨如何在“健康中国”理念下助力中国罕见病事业的发展。

夏尔亚太区负责人及代理中国区总经理方思璞联合罕见病发展中心创始人兼主任黄如方
共同致力推进罕见病事业发展
创新为源,研发创新医疗解决方案
目前,全球罕见病患者总人数高达3.5亿,其中中国罕见病患者预计超过1600万人。由于缺乏认知和有效的治疗方案,罕见病患者长期面临诊断率低、治疗率低两大难题。在已知的7000余种罕见病中,仅5%有药可医,30%的患儿寿命不超过5周岁。
在中国,诊疗率双低的问题显得尤为突出。以血友病为例,全国13.6万血友病患者中,诊断率不到10%;而在认知度更低的溶酶体贮积症领域,法布雷病诊断率低于5%,戈谢病的诊断率为9%且半数患者在初诊时不能被诊断出,或被误诊为其他血液疾病。
面对全球罕见病患者未被满足的迫切医疗需求,夏尔亚太区医学事务部负责人刘森旺博士就罕见病药物创新研发主题与业界同仁进行深入探讨,分享了夏尔在全球罕见病领域的经验与实践。他表示:“目前,夏尔正不断加大在研发领域的投入,努力推动全球40个临床产品项目的发展,其中约70%的临床项目针对罕见病适应证,50%以上的项目已处于三期临床或上市注册阶段。秉承‘创新为源、患者至上’的理念,夏尔一直致力于通过持续推进罕见病领域的科技突破,推动罕见病产品线的不断丰富,为患者群体提供帮助。”

夏尔亚太区医学事务部负责人刘森旺博士在大会开幕式致辞
目前,夏尔在全球超过40种上市产品中,76%的产品针对罕见病领域,重点关注免疫疾病、血液疾病、遗传疾病、肠胃疾病和内科疾病等。借助精准创新和技术,夏尔首创突破性创新疗法,使用单克隆抗体技术和基因技术分别针对遗传性血管性水肿(HAE)和血友病研发新的治疗方法,这些突破性的创新疗法,将有力地填补相关治疗领域的空白,更好地解决患者亟待满足的需求。
患者至上,多方通力协作探寻发展之路
在本届论坛上,夏尔邀请多位国内外专家学者、临床医生、患者组织代表等专业人士,共同探讨并展望罕见病领域的现状及发展前景,重点关注以法布雷病、戈谢病为代表的溶酶体贮积症(LSD)在中国未被满足的诊疗需求,助力推进疾病诊疗水平、为LSD患者发声、提升医疗可及性。
LSD的发病机制尚处于探索阶段,在诊断和药品研发领域都面临着较多挑战。以法布雷病为例,该疾病具有潜在致命性,男性患者平均生存期较健康人群缩短20年,女性患者平均生存期则缩短约10年,但许多医生却对其知之甚少。患者通常要经历一段漫长的求医问诊之路,尤其是在诊断初期,常因被漏诊、误诊为其他疾病而延误治疗,平均确诊需12年,严重影响患者的生命质量。
北京协和医院儿科教授魏珉表示:“溶酶体贮积症(LSDs)在中国面临疾病认知率低、诊疗率低等问题。本次大会为我们创造了一个高附加值的交流平台,希望通过各方携手努力,助力加强医护人员、患者和社会公众对疾病的认知,加速国际前沿治疗解决方案的引进,以期提高诊疗率,改善中国LSD患者的状况。”

专家及患者组织代表在夏尔卫星会上宣誓共迎LSD挑战
携手合作,助推中国罕见病事业发展
今年是中国改革开放40周年,随着中国医疗改革步伐的不断深化,当下中国罕见病行业的发展迎来黄金发展时期。 近期中国政府一系列利好政策与举措相继出台——发布《第一批罕见病目录》、提速罕见病药品评审、鼓励罕见病创新药物研发等,将有利于吸引越来越多的相关方积极投身中国罕见病事业的发展。
夏尔亚太区负责人及代理中国区总经理方思璞(Peter Fang)先生表示:未来五年,夏尔将继续与以罕见病发展中心为代表的各方合作伙伴通力合作,继续持续努力把更多突破性的创新医疗解决方案引进中国市场,为中国1600万罕见病患者带来生命曙光。”
访谈风采
溶酶体贮积症是一组遗传性代谢疾病,是由于基因突变致溶酶体中有关酸性水解酶缺陷,导致机体中相应的生物大分子不能正常降解而在溶酶体中贮积,引起细胞组织器官功能的障碍。既可导致多种系统的病变,也可仅限于神经系统,自出生至成年期均可发病。记者针对溶酶体贮积症采访了北京协和医院儿科魏珉教授、上海瑞金医院肾脏科副主任医师潘晓霞教授和上海市儿童医院血液肿瘤科主治医师陈凯。
提问:溶酶体贮积症的遗传原理是什么?疾病携带者如结婚生育,在疾病筛查方面有什么建议?
魏珉教授:溶酶体贮积症有常染色体隐性遗传的,有伴性遗传的,最根本的还是要先确诊,确诊之后比如是常染色体隐性遗传性疾病,大夫就知道父母再次生育,会有四分之一的概率再生出患儿,四分之二概率生出携带者,四分之一的概率生出健康的孩子。有了这类疾病患者的家庭就可以称为高危家庭。对于高危家庭成员,如果准备结婚生育,目前可以做到产前诊断,或针对家庭中已有先证者的这个疾病进行携带者筛查。只要先证者得到了确诊,医生就可以明确了解其遗传规律。
提问:对溶酶体贮积症这类疾病的防治目前在中国面临着哪些挑战?
魏珉教授:最大的挑战还在于临床医生得认识这个病,诊断是一切治疗的基础。医生们需要了解这些罕见病的临床表现是什么,只有知道了才有可能转诊给相应专家去确诊。举个例子,今天讲的戈谢病有肝脾肿大的特征,但是很多常见疾病或遗传代谢病都可以有肝脾肿大,尤其遗传病多数非常罕见,医生不可能每一个都熟悉,所以在这一步就会出现漏诊,这是难点。至于诊断方面,酶学诊断和基因诊断不是特别难的问题,我国目前起码有四个医院都可以诊断。比如,北京协和医院自上世纪80年代起就已经开始做酶学诊断。从2000~2017年,协和医院在这17年中诊断了溶酶体贮积症1605例,其中产前诊断是643例,约占到总体的40%。
提问:溶酶体贮积症患者目前推荐的治疗方案是什么?
潘晓霞教授:我先说一下法布雷病,它的特异性治疗在国际上很早就有了,即酶替代治疗(ERT),早在十多年前就在国际上市,它的安全性、疗效也得到了大家的肯定。我从2000年左右开始致力于研究法布雷病,我们也希望通过媒体的呼吁,和政府、药企,包括患者群体一起,尽快共同努力让这些药进入临床,让患者能够用上,满足患者的用药需求。
魏珉教授:我也关注这个问题很多年了,确实在国外已经有替代治疗的方法。比如戈谢病,一个国外的基金会将相关治疗药物赠送给中国患者,已经在中国治疗了近100多个患者,患者治疗了以后确实可以看到有很明显的效果。这个酶替代疗法(ERT)在世界范围内已经有了,但是还未进入到中国。我希望在药物引进的审批过程中能进一步加速。
提问:溶酶体贮积症有黄金治疗期吗?
潘晓霞教授:法布雷病一旦治疗就是终身治疗。采用ERT疗法,在国际上实行的准则是——如果是16岁以上的男性,一旦确诊就建议治疗,且需要终身治疗。而如果是女性携带者,如果症状很明显,比如出现蛋白尿,就建议接受治疗。
陈凯医师:戈谢病是终身治疗,尤其是Ⅰ型的,而ERT是目前最推荐的疗法。比如,有些患儿可能脾肿胀很厉害,我们采取治疗措施后,一般3~6个月后它就会慢慢缩小,坚持用药2~3年,脾就能恢复正常,但必须坚持终身用药,否则一旦停药后就会复发。而复发后的重新治疗很可能会花费更大的代价和时间。所以,适时选择ERT会获得比较好的预后,让患者可以像正常人一样生活、结婚、上学和工作。
魏珉教授:有一个很好的案例,戈谢病治疗十周年时,曾经开了一个患者会,其中有一个患者给我的印象非常深刻。他由于在基金会的赞助下较早地进行了酶替代治疗,治疗后不仅上了大学,还考取了研究生。在大会上,他全程用英文演说自己的心路历程,而对比他之前生病时的照片,你根本看不出来他曾经是患者,现在完全像一个正常人。这让大家都非常感动,让我们做医生的觉得所有的努力都是值得的。
提问:如果确诊前已做其他治疗,会影响确诊后治疗吗?
潘晓霞教授:法布雷病罕见,常不为许多临床医师所认识,因此漏误诊率非常高。在未确诊前,很多患者会误诊为其他疾病而给予一些非针对性的治疗,不仅延误病情,反之可能因为药物的毒副作用而增加患者的痛苦。比如部分出现肾脏损害的患者,可能误诊为原发性肾脏病,如果这些患者出现大量蛋白尿,临床可能会给予激素、免疫抑制剂治疗,这些药物对法布雷病来说没有太大效果,但是这些药物有很大副作用,比如长期应用激素可造成股骨头坏死,严重者需要置换股骨头。其他药物可能导致其他的毒副作用,并会影响生活质量。法布雷病的治疗建议尽早开始,而延误诊断的结果导致许多患者在疾病后期出现了严重脏器损害的情况下方被发现,此时器官的损害往往不可逆了,而药物的治疗效果也会差。所以,非针对性治疗可能引发许多的毒副作用从而影响患者的生活质量,而延误诊断导致未及时给予针对性治疗可能让患者丧失最佳的治疗时间。
提问:国外在溶酶体贮积症患者的诊疗方面是否有值得借鉴之处?
魏珉教授:国外对于罕见病有一套成熟的管理措施,所有的罕见病都是注册的,只要是罕见病,一般情况下国家会支持治疗,观察的过程也有一个团队,除了有医学专业的,还有饮食、运动方面。比如说溶酶体贮积症有的涉及到耳鼻喉、骨科、外科的问题,都由团队来管理病人,我觉得这是咱们将来努力的方向。值得高兴的是,咱们国家已经迈出了第一步,公布了第一批罕见病目录,承认这样一个群体。除了罕见病目录之外,国家也发布了加速境外已上市罕见病药品审批的相关政策,患者未来将有机会获得创新药物。
提问:现在是互联网大数据时代,有些医院已经有了自己的罕见病数据分析,全国医院间是不是会互相合作,互通这类数据?
潘晓霞教授:最近好像协和医院在做全国罕见病的登记,可能将来会形成一个大数据,而我们现在还是各个中心收集自己的资料,没有形成一个网络。
提问:溶酶体贮积症在我国哪些医院可以就诊?
魏珉教授:一般而言,在北京有协和医院、北大医院和儿童医院,上海有瑞金医院或是各类儿童医院。此外,就儿科而言,还有致力于儿科遗传方面研究的武汉同济,还有像广东妇幼保健院等。
发表评论
最新评论

-

new如“悦”而至 中国“达”案,鼻部疾病靶向生物治疗学术研讨会于成都举办
2025-05-19 -

new院内外同治 多学科协同控糖,国际前沿新技术iCGM助力糖尿病管理
2025-05-19 -
04-292025
高危膀胱癌患者联合治疗方案有新突破
-
04-262025
晚期黑色素瘤一线免疫疗法有新方案
-

独辟蹊径!易俊林团队发现缩小鼻咽癌患者放疗靶区新方法
2024-03-10 -

《2023中国高脂血症诊疗现状与疾病管理调研报告》在第六届进博会发布
2023-11-07 -

传承百年创新征程,攻坚糖尿病未竟之业
2023-04-12 -
18厘米超长阑尾被保住!《胃肠内镜》刊登我国专家保阑尾ERAT治愈最长阑尾急腹症病例
2022-12-11 -

泛肿瘤TRK抑制剂罗圣全®在中国获批,开启个体化医疗新篇章
2022-07-29
-

聚焦世界脑健康日五大核心目标,携手同行共促脑健康,2023中国脑健康大会成功举办
2023-07-27 -
18厘米超长阑尾被保住!《胃肠内镜》刊登我国专家保阑尾ERAT治愈最长阑尾急腹症病例
2022-12-11 -

全球首部钙调磷酸酶抑制剂治疗风湿免疫性疾病临床应用专家共识全国发布
2023-06-12 -

县域医疗机构消化专科精准能力提升项目湖南试点启动会在长沙召开
2023-05-30 -
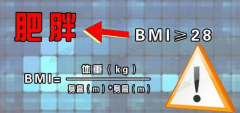
【5·11世界防治肥胖日】今天,大声喊出“我要瘦”!
2016-05-17 -

两院院士增选工作正式启动!医学领域儿科、麻醉、神经病学优先增选推荐!
2023-07-27 -

长期肥胖增加食管癌和贲门癌风险
2017-08-06



